Identification of spatially associated subpopulations by combining scrNAseq and sequential fluorescence in situ hybridization data.
Abstract
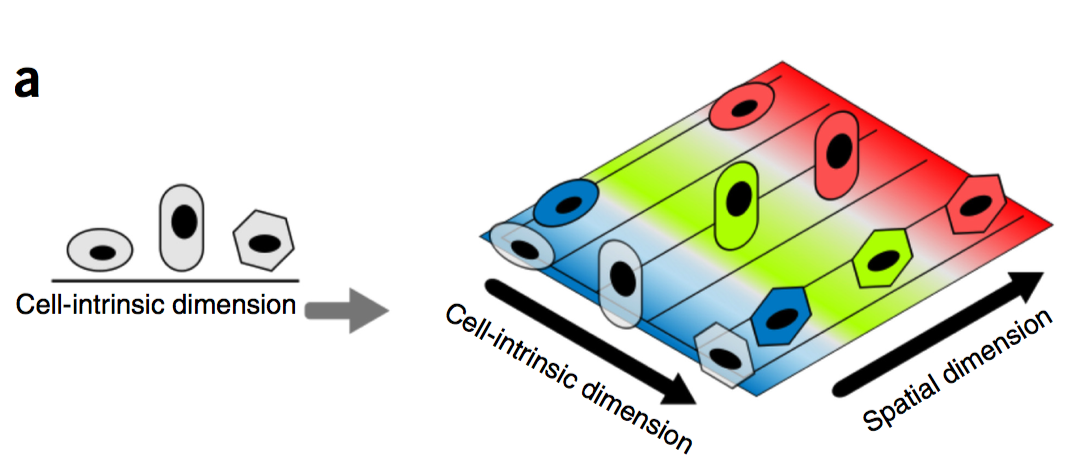
How intrinsic gene-regulatory networks interact with a cell’s spatial environment to define its identity remains poorly understood. We developed an approach to distinguish between intrinsic and extrinsic effects on global gene expression by integrating analysis of sequencing-based and imaging-based single-cell transcriptomic profiles, using cross-platform cell type mapping combined with a hidden Markov random field model. We applied this approach to dissect the cell-type- and spatial-domain-associated heterogeneity in the mouse visual cortex region. Our analysis identified distinct spatially associated, cell-type-independent signatures in the glutamatergic and astrocyte cell compartments. Using these signatures to analyze single-cell RNA sequencing data, we identified previously unknown spatially associated subpopulations, which were validated by comparison with anatomical structures and Allen Brain Atlas images.
Contribution: Processing of high-throughput datasets. Data analysis and interpretation. Writing and editing of manuscript.